Weight-of-Evidence Assessments: Unpacking New Guidance on Carcinogenicity Testing
On November 1, 2022, the U.S. Food and Drug Administration (FDA) adopted an addendum to the guidance titled “S1B(R1) Testing for Carcinogenicity of Pharmaceuticals,” which had previously been finalized by the International Council for Harmonization (ICH). The guidance document and integrated addendum detail an integrative, weight-of-evidence (WoE) method of determining whether carcinogenicity testing in rats will be informative in human risk assessments for drug approvals.
This blog post will explain how the WoE approach is used in toxicology studies and how the S1B(R1) addendum could affect your nonclinical program.
Background
The addendum’s development was informed by both prospective and retrospective evaluations of animal carcinogenicity data submitted to the ICH.1,2,3 Its appendix offers case studies on the application of WoE in determining the utility of 2-year rat studies. Of note, this guidance is specific to small molecule pharmaceuticals; similar guidance for biologics can be found in the ICH’s S6(R1) Preclinical Safety Evaluation for Biotechnology-Derived Pharmaceuticals.
Weight-of-evidence approach
The WoE approach has long been used in toxicological safety assessments for drugs, general chemicals, food additives, pesticides, and more. FDA guidance on nonclinical safety testing for pediatric products has an informative explanation of what a WoE approach entails:
An approach that evaluates information from several sources to decide if there is sufficient evidence to support the development of pharmaceuticals for pediatric use or whether additional nonclinical testing is warranted to address potential safety concerns. The weight given to the available evidence depends on factors such as the quality of the data, consistency of results, nature and severity of effects, and relevance of the information. The weight of evidence approach requires use of scientific judgment and, therefore, should consider the robustness and reliability of the different data sources.
In short, a WoE approach to safety assessment integrates information from multiple different sources – weighing the quality, consistency, severity, and human relevance of the data. The term “weight of evidence” appears 18 times in FDA guidance documents and is primarily applied when assessing developmental and reproductive toxicity, immunotoxicity, genotoxicity, pediatric safety, and carcinogenicity.
WoE assessments are especially useful and routinely leveraged in 505(b)(2) new drug applications (NDAs), since there is often a large dataset of safety information on the active pharmaceutical ingredient (API) from the Listed Drug (LD). However, the quality and relevance of that data can vary.
For example, for a combination product including one or more already-approved drugs, the WoE surrounding potential pharmacodynamic or toxicological interactions must be assessed, to determine whether the combination can be tested in clinical trials without a nonclinical toxicity study.
Another example may be the development of a new chronic use drug using a different route of administration from the LD (for instance, a topically administered product based on an orally administered LD). Here, a carcinogenicity study using the different route of administration may not be necessary if there were no alerts in the chronic study (e.g., no pre-neoplastic lesions noted in a 9-month topical minipig study) and no carcinogenicity concerns in the original oral carcinogenicity studies.
The S1B(R2) Approach
The WoE assessment described in the updated S1B(R2) addendum evaluates if a two-year rat study will provide value for human carcinogenicity assessment. It begins with collecting all relevant information from in vitro, in vivo, or in silico studies, either sponsor-conducted trials or the literature. The information should be relevant according to six key WoE factors: target biology, secondary pharmacology, previously conducted studies (e.g., chronic studies or transgenic mouse studies), hormonal effects, genotoxicity, and immune modulation (Figure 1). Pharmacokinetic and systemic exposure data is also important to consider.
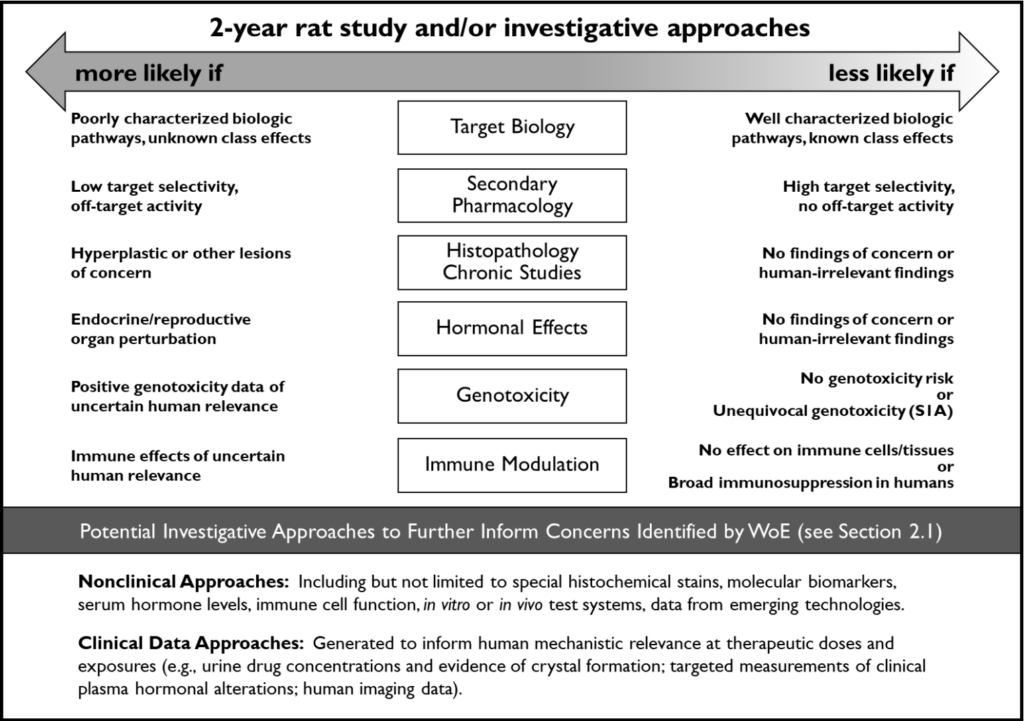 Figure 1. Integration of key WoE factors and potential investigative approaches to further inform on the value of conducting a 2-year rat study for assessment of human carcinogenic risk. Source: ICH S1B(R1), August 2022.
Figure 1. Integration of key WoE factors and potential investigative approaches to further inform on the value of conducting a 2-year rat study for assessment of human carcinogenic risk. Source: ICH S1B(R1), August 2022.
An expert assessment of the totality of information within each WoE factor should lead to one of three possible conclusions:
| Carcinogenic potential in humans is likely | Document WoE assessment, and seek regulatory agreement that a two-year rat study and/or mouse study will not provide value. |
| Carcinogenic potential in humans is unlikely | Document WoE assessment, and seek regulatory agreement that a two-year rat study will not provide value. A transgenic mouse study may still be needed, though there are cases where no carcinogenicity testing is required. |
| Carcinogenic potential in humans is uncertain | Conduct the two-year rat carcinogenicity study and possibly additional in vivo carcinogenicity studies. |
What about the mouse carcinogenicity study?
Carcinogenicity studies in mice, particularly transgenic mice, are a recommended component of a carcinogenicity assessment, even if the WoE assessment determines that a two-year rat study would not provide value. This is because the transgenic mouse model provides an alternative model for carcinogenesis and thus provides value in understanding human carcinogenic risk. However, a mouse study may not be informative if the drug product is likely to be a human carcinogen, or if there is little carcinogenic risk in humans and exposures in mice cannot approximate human exposures. A transgenic mouse study does not need to be completed before a WoE assessment.
Takeaway
The S1B(R1) addendum outlines a WoE approach for determining if carcinogenicity studies in rats will be informative to the human carcinogenicity risk assessment for small molecule pharmaceuticals. This addendum highlights an important shift in carcinogenic potential assessments for drug products, where the need for additional carcinogenicity studies is evaluated, rather than assumed.
Using a WoE approach may reduce the size of a nonclinical development program, saving time and cost and reducing the number of animals used. In the case of 505(b)(2) NDAs where carcinogenicity information for the drug substance is leveraged from the LD, carcinogenicity studies with the new drug product are often waived, even for a change in the route of administration. It is important to interact with regulatory bodies throughout development to understand their perspectives and gain agreement on which nonclinical studies are needed.
At Premier Consulting, our toxicologists have deep expertise in the strategic design and execution of nonclinical programs for all regulatory approval pathways. Contact us to find out how they can help you develop and apply WoE toxicological assessments to your development plan.
Author:
Madelyn “Mimi” Huang, PhD
Toxicologist
References:
- Proposed Change to Rodent Carcinogenicity Testing of Pharmaceuticals – Regulatory Notice Document. ICH, 2016. URL: https://database.ich.org/sites/default/files/S1%28R1%29_EWG_RND.pdf
- The ICHS1 Regulatory Testing Paradigm of Carcinogenicity in Rats: Status Report 2021. ICH, 2021. URL: https://database.ich.org/sites/default/files/S1_StatusReport_2021_0823.pdf
- Hisada S, Tsubota K, Inoue K, Yamada H, Ikeda T, Sistare FD. Survey of tumorigenic sensitivity in 6-month rasH2-Tg mice studies compared with 2-year rodent assays. J Toxicol Pathol. 2022 Jan;35(1):53-73. doi: 10.1293/tox.2021-0031. Epub 2021 Nov 1. PMID: 35221496; PMCID: PMC8828610.