The FDA’s Technical Rejection Criteria for Study Data: Does Your eCTD Submission Comply?
When a submission is sent through the FDA’s Electronic Submissions Gateway, it goes through an automated check of many validation rules as specified in the Electronic Common Technical Document (eCTD) submission standards guidance. This validation check determines if the submission is uploaded to the FDA’s system or is technically rejected.
To help sponsors understand how the FDA uses the eCTD technical validation rules to verify conformance, the agency developed the technical rejection criteria (TRC) for study data and began enforcing the TRC for study data in September 2021.
What are the relevant technical rejection criteria documents?
The FDA has published three binding guidance documents regarding the TRC. The first two were published in 2014:
- Providing Regulatory Submissions in Electronic Format — Submissions Under Section 745A(a) of the Federal Food, Drug, and Cosmetic Act mandates that NDAs, ANDAs, and INDs be submitted in electronic format
- Providing Regulatory Submissions in Electronic Format — Standardized Study Data requires that study data be submitted in an electronic format that the FDA can process, review, and archive
Beginning 24 months (for NDAs, ANDAs, and BLAs) or 36 months (for commercial INDs) after the publication of these guidance documents, the FDA started being able to issue a refuse to file (RTF) letter for NDAs and BLAs, or a refuse to receive (RTR) letter for ANDAs for non-conforming submissions.
A final binding guidance, Providing Regulatory Submissions in Electronic Format — Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications, published in April 2017, required that electronic submissions be formatted according to eCTD submission standards and defined technical rejection criteria.
The publication of the Data Standards Catalog (DSC) followed. The DSC lists the required data standards, accepted data formats and versions, and implementation dates for studies starting on or after December 17, 2016 (for NDAs, ANDAs, and BLAs), and December 17, 2017 (for INDs). Sponsors whose studies started after these dates must submit data in the formats supported by the FDA and listed in the DSC.
The FDA also issued the Study Data Technical Conformance Guide, which contains detailed information on how to meet the requirements contained in the DSC.
Where are the technical rejection criteria for study data applied?
The TRC for study data apply only to documents submitted with the metadata of “study report,” “preclinical study report,” and “legacy study report” in the Module 4 and Module 5 sections listed below:
- 4.2.3.1: Single-Dose Toxicity
- 4.2.3.2: Repeat-Dose Toxicity
- 4.2.3.4: Carcinogenicity
- 5.3.1.1: Bioavailability Study Reports
- 5.3.1.2: Comparative BA and BE Study Reports
- 5.3.3.1: Healthy Subject PK and Initial Tolerability Study Reports
- 5.3.3.2: Patient PK and Initial Tolerability Study Reports
- 5.3.3.3: Intrinsic Factor PK Study Reports
- 5.3.4.1: Healthy Subject PD and PK/PD Study Reports
- 5.3.4.2: Patient PD and PK/PD Study Reports
- 5.3.5.1: Study Reports of Controlled Clinical Studies Pertinent to the Claimed Indication
- 5.3.5.2: Study Reports of Uncontrolled Clinical Studies
What are the technical rejection criteria for study data?
The TRC are dependent on study start date, application type, study type, and FDA center. The ts.xpt dataset is the mechanism used to determine which versions of the data standard listed in the DSC are required, based on the study start date. Therefore, regardless of the start date, if a study report is submitted in the eCTD sections covered by the TRC, a ts.xpt file must be included. In cases wherein datasets are not required, a simplified ts.xpt file is included.
There are four HIGH-severity TRC validation codes for study data (Table 1):
Table 1
| Validation Code | Criteria Being Verified |
| Code 1734 | A trial summary dataset (ts.xpt) containing the study start date in YYYY-MM-DD format is present. |
| Code 1735 | The correct STF file tags are used for datasets and corresponding data definition files. |
| Code 1736 | · For Standard for the Exchange of Nonclinical Data study data, a demographic dataset (dm.xpt) and define.xml were submitted
· For Study Data Tabulation Model study data, a DM dataset (dm.xpt) and define.xml were submitted · For Analysis Data Model study data, a subject-level analysis dataset (adsl.xpt) was submitted |
| Code 1789 | For all Module 4 and Module 5 sections (except sections 4.3, 5.2, 5.3.6, and 5.4), a file was submitted without an STF file. |
Table 2 illustrates how these codes are applied.
Table 2
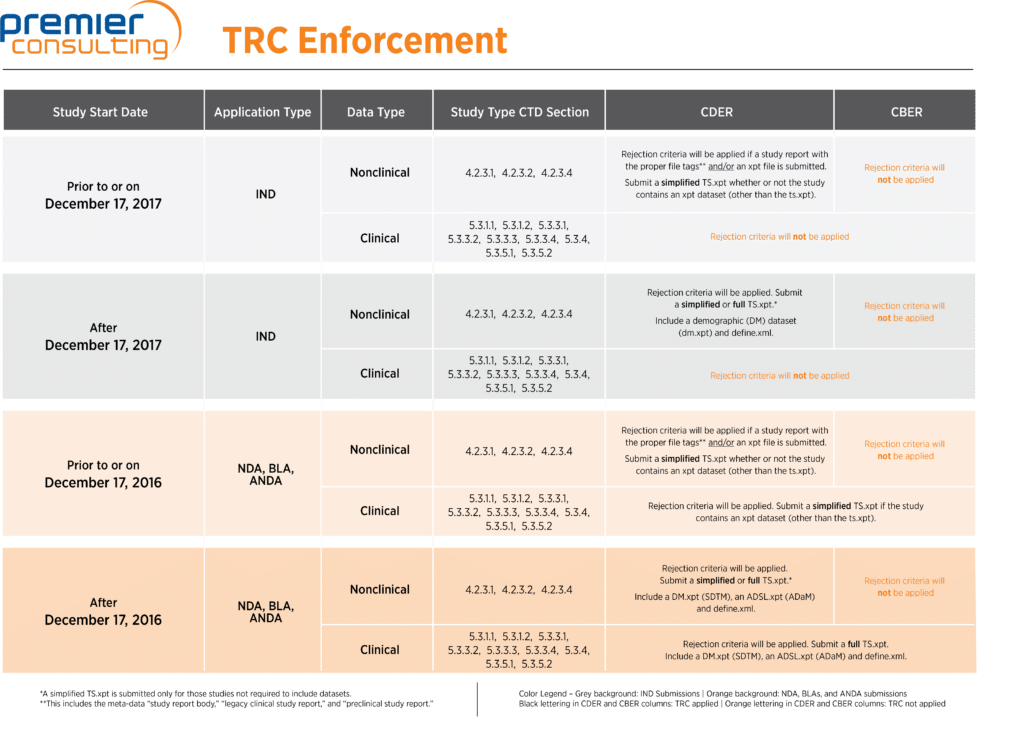
How are the study data standards enforced?
The automated process for the TRC validation check for study data is performed sequentially, starting with code 1789 and then moving through codes 1734, 1735, and 1736. The process moves to the next code check only if it passes the previous one. As soon as a submission fails a code check, validation stops; the submitter is notified via an Electronic Submissions Gateway acknowledgment that the submission was technically rejected and was therefore not uploaded into the FDA system. The submission must be corrected and sent through the gateway again. When technical validation passes, the submission is uploaded into the FDA system and made available to the review division.
When should study data standards be incorporated?
The FDA’s Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research recommend that IND sponsors and NDA applicants consider implementing study data standards as early as possible in the product development life cycle so that data standards are accounted for in the design, conduct, and analysis of studies. Proposed data standards can be discussed in a pre-IND meeting.
The FDA also recommends including a Study Data Standardization Plan (SDSP) at the time of the IND filing to document how data standards will be applied for a program’s completed and planned studies, and updating it periodically. An SDSP is typically presented within an IND’s general investigational plan, and the latest version should be provided to the FDA in a pre-NDA briefing package.
To help sponsors navigate the TRC for study data, the FDA created a TRC Self-Check Worksheet. Sponsors also can request a Type C meeting to discuss technical concerns or questions.
Premier Consulting’s philosophy of beginning with the end in mind provides a sound pathway to success. Our deep expertise with electronic submissions can help ensure that your submissions are compliant with all FDA eCTD specifications. Contact us to find out how.
Author:
Annette F. Arlinghaus
Director, Regulatory Operations
References:
- Study Data Standards Resources
- Data Standards Catalog
- Study Data Technical Conformance Guide
- Study Data for Submission to CDER and CBER