Risk Evaluation and Mitigation Strategies (REMS) Basics
The Food and Drug Administration is responsible for ensuring that human drugs are safe and effective, while also advancing public health by helping to speed product innovations. In determining if a drug should be marketed, the Agency must weigh the benefits of the therapy against its potential risks to the patient. If a drug or biologic product has serious safety risks, the FDA can mandate that these risks be mitigated and/or evaluated before or after marketing approval.
REMS Basics
In 2007, the Food and Drug Administration Amendments Act (FDAAA) and one of its provisions gave FDA the authority to require Risk Evaluation and Mitigation Strategies (REMS) from manufacturers to allow a drug or biological product to be used in a way that also ensures that its benefits outweigh its risks (see the REMS draft guidance). This means that when safety risks are identified (either before or after marketing approval of the product), the FDA can mandate certain REMS elements with which the Sponsor must comply to ensure that the benefit to risk ratio is acceptable and safety concerns are minimized. These requirements are in addition to the professional labeling of the product, and can include any or all of the following for an ANDA, NDA, or BLA:
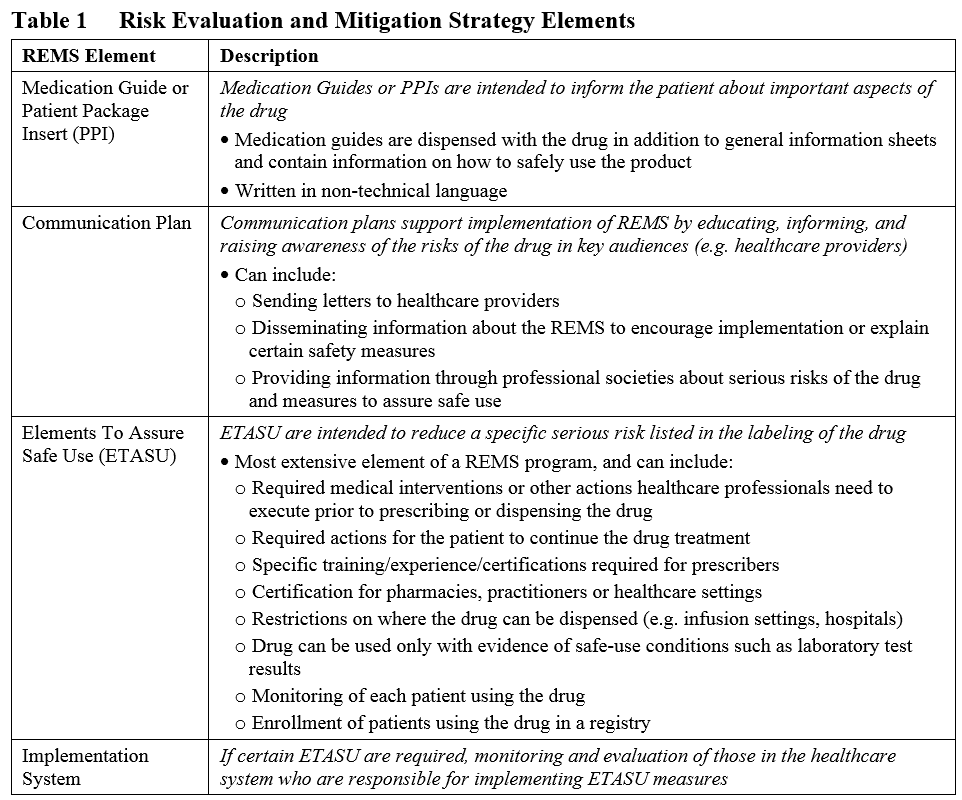
All REMS must include a timetable for assessing the effectiveness of their safety measures. This timetable must be at least as frequent as 1.5, 3, and 7 years after the REMS is approved. If the assessments indicate that changes are needed or that the REMS has met its goals, the REMS requirement may be modified or even eliminated after 3 years (see the FDA guidance on REMS modifications and revisions). The following are examples of assessments of REMS effectiveness:
- Survey data describing healthcare professionals’ understanding regarding the safe use of the drug
- Summaries of adverse events associated with the drug that REMS was designed to address
- Measures of prescriber compliance with certification and REMS requirements (e.g. training/enrollment procedures, completion of patient baseline form, complying with discontinuation procedures)
- Data on the types of patients using the drug and conditions under which the drug is used
- Number and percentage of patients monitored for potential serious adverse events during drug treatment
A list of REMS that are currently approved for various drugs or drug classes can be found on the REMS@FDA website.
Case Study: REMS for Opioids
Because of recent focus on prescription drug abuse of opioid products, one of the most publicized REMS programs is for ER/LA opioid analgesic products which began in 2012 and currently includes 65 NDA and ANDA products (the list of products can be found on the ER/LA opioid analgesics REMS FDA website). This REMS program requires products to provide a medication guide and adhere to ETASU, including mandated prescriber training and evaluation of training compliance.
In May of 2016, a joint meeting of the Drug Safety and Risk Management (DSaRM) Advisory Committee and the Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC) was held to assess the ER/LA opioid analgesics REMS program and determine whether revisions or modifications should be made. The results from the ETASU indicated that while the number of prescriptions of ER/LA opioid analgesics has declined slightly to 21.2 M in 2014 from 22.3 M in 2010, the number of prescribers who have completed REMS-compliant continuing education training was 53% lower than the goal set for 2015.
Participants at the meeting discussed various ways to improve compliance with the training goal, but final recommendations have yet to be instituted in the REMS. While not every periodic review of a REMS program will be associated with an advisory committee meeting, this example illustrates the agency’s commitment to using REMS to critically evaluate the risks and benefits of a drug and provide an avenue for continuing to improve safe use.
REMS and 505(b)(2) Product Development
For a Sponsor submitting a New Drug Application via the 505(b)(2) pathway that relies in part on the safety or effectiveness of a listed drug, the REMS status of the listed drug can influence the REMS requirement for the proposed product.
If the listed drug already has REMS requirements, a Sponsor relying on that listed drug for their 505(b)(2) NDA will likely be subject to the same requirements. In this case, it makes sense to include the REMS proposal for the product as part of the NDA submission to avoid delays in the review process.
Premier Consulting has experience in assessing the REMS requirements for various drugs and determining the most efficient strategy to meet these regulatory mandates. Please contact us for more information.