Preclinical and Nonclinical Studies—What Is the Difference, and Where in Your Program Should They Fall?
Have you ever wondered about the difference between preclinical and nonclinical studies? While those in scientific fields like pharmaceutical development are accustomed to words with specific, agreed-upon definitions to facilitate communication and minimize misunderstandings, these terms seem to have slipped through the cracks. Their similar meanings often make it difficult to determine which word is appropriate in a given context and which studies fit their definitions across the development landscape.
When should each term be used, and what are the appropriate timelines for preclinical and nonclinical studies within a drug development program?
When “preclinical” and “nonclinical” are used
Preclinical refers to studies occurring prior to clinical testing, while nonclinical refers to studies not related to, involving, or concerned with the direct observation and treatment of living patients. Attempting to differentiate the words raises a few questions:
- Which of the two, if either, includes in vitro studies?
- Which includes efficacy studies in animal models, and which includes toxicology programs?
- Are these terms limited to studies required before the first-in-man clinical study, or do they also include reproductive toxicology and chronic studies usually conducted after an IND is opened?
Regulatory agencies do not seem to make a strong distinction in usage of the terms. The European Medicines Agency uses either word in guideline titles and uses them interchangeably within the text. Meanwhile, the Food and Drug Administration specifically chooses to use the word nonclinical with a disclaimer that these studies are “often referred to as preclinical studies when conducted before first-in-human clinical studies.”
The FDA offers a more substantive definition of nonclinical laboratory studies in Section 58.3 of Good Laboratory Practice for Nonclinical Laboratory Studies:
Nonclinical laboratory study means in vivo or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety. The term does not include studies utilizing human subjects or clinical studies or field trials in animals. The term does not include basic exploratory studies carried out to determine whether a test article has any potential utility or to determine physical or chemical characteristics of a test article.
Many pharma and biotech companies identify themselves as being in the preclinical phase of development if their compounds have not yet been tested in humans. Meanwhile, many service providers use nonclinical to describe their animal lab capabilities to be inclusive of studies that are done after an IND filing, such as reproductive or chronic toxicology. Since the two study types are so similar and overlap heavily, the words are often used interchangeably, and it would not be incorrect to use either to describe IND-enabling studies.
What nonclinical/preclinical studies are needed, and when?
It goes without saying that every drug development program is unique and requires regulatory input early and often. Premier Consulting’s strength lies in providing customized development and regulatory strategies for complex programs. Regulatory guidelines outline, in general terms, the optimal times to perform required studies.
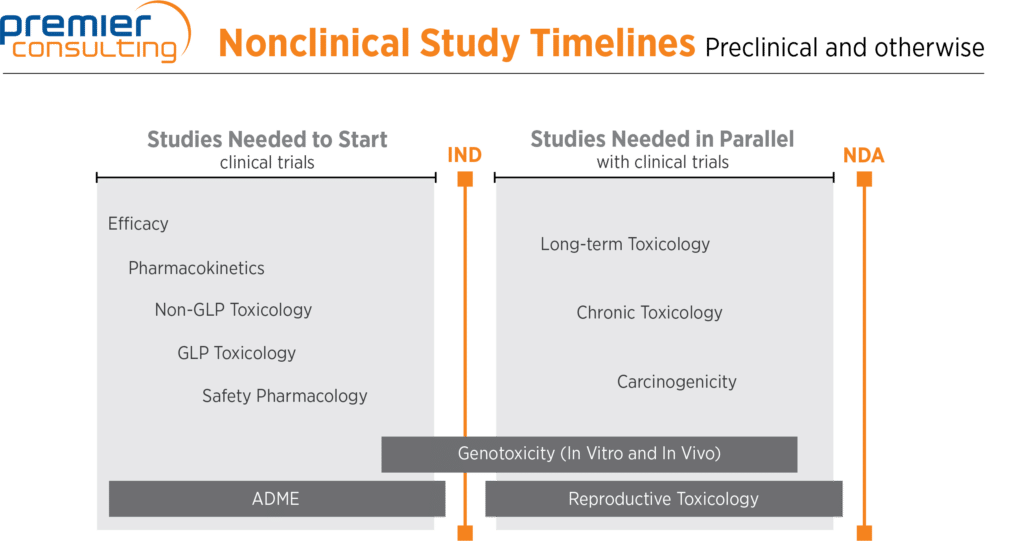
In standard NCE or NME programs:
- For a non-oncology new chemical entity (NCE) or new molecular entity (NME) program, efficacy studies and pharmacokinetics studies typically take place first
- Then come non-GLP and GLP toxicology studies (typically rodent and non-rodent) and safety pharmacology (central nervous system and respiratory studies typically conducted in rodents, and cardiovascular studies typically conducted in non-rodents)
- A subset of genotoxicity studies must be started before an IND is filed (in vitro mutagenesis and chromosomal aberration studies), and the remainder must be completed before Phase 2 of clinical trials (in vivo micronucleus studies typically conducted in rodents)
- Long-term and chronic toxicology studies, carcinogenicity studies (typically two years long in rodents), and reproductive studies (fertility, teratology, and pre- and postnatal studies) must take place before NDA filing
- Absorption, distribution, metabolism, and excretion studies are sometimes required throughout the drug development process, but are needed mostly at the beginning
Other programs: Oncology and biologics
When developing oncology NCEs, sponsors can include safety pharmacology as endpoints in GLP studies, and no standalone studies are required. In addition, long-term, chronic, and some reproductive toxicology studies may not be needed, and carcinogenicity studies can be skipped as well. Lastly, genetic toxicology studies can be performed later in an oncology NCE program, as they are not needed until the marketing application is developed.
For biologics, immunological endpoints such as anti-drug antibodies are added for many of the nonclinical studies. Rodents are not used as frequently as in NCE programs; in fact, the species selection is an integral part of the early meetings with regulatory agencies for biologics programs. Genetic toxicology studies are not required, and carcinogenicity studies are not always needed. Oncology biologics programs are similar to oncology NCE programs, but they do not include genetic toxicology studies.
Contact us to discover how Premier Consulting can provide strategy and regulatory support for your nonclinical program and minimize risk during its execution through Premier Partners. Our cross-functional team is fully equipped to advance your development program as you prepare to open an IND and beyond.