Is Your In Vitro Diagnostic Exempt from Investigational Device Exemption Regulations?
An investigational device exemption (IDE) allows an investigational device to be used in a clinical study to collect safety and effectiveness data.[1] However, if certain criteria are met, many in vitro diagnostic (IVD) devices are released from IDE regulations. This blog post discusses how device studies are classified and explores scenarios where IVDs qualify for exclusion from IDE regulations.
Categorizing device studies according to IDE regulations
According to the IDE regulations under 21 Code of Federal Regulations (CFR) 812, device studies are categorized into three types:
- Significant risk (SR) device studies, which require an approved IDE. Generally, device studies are considered a significant risk if the device is invasive, used to support or sustain life, intended to be the sole basis of diagnosis or decision-making, or associated with a potential for serious risk to the subject (see Figure 1).
- Nonsignificant risk (NSR) device studies, which must meet the abbreviated IDE requirements outlined in 21 CFR 812.2(b)
- Exempt studies that do not require an IDE.
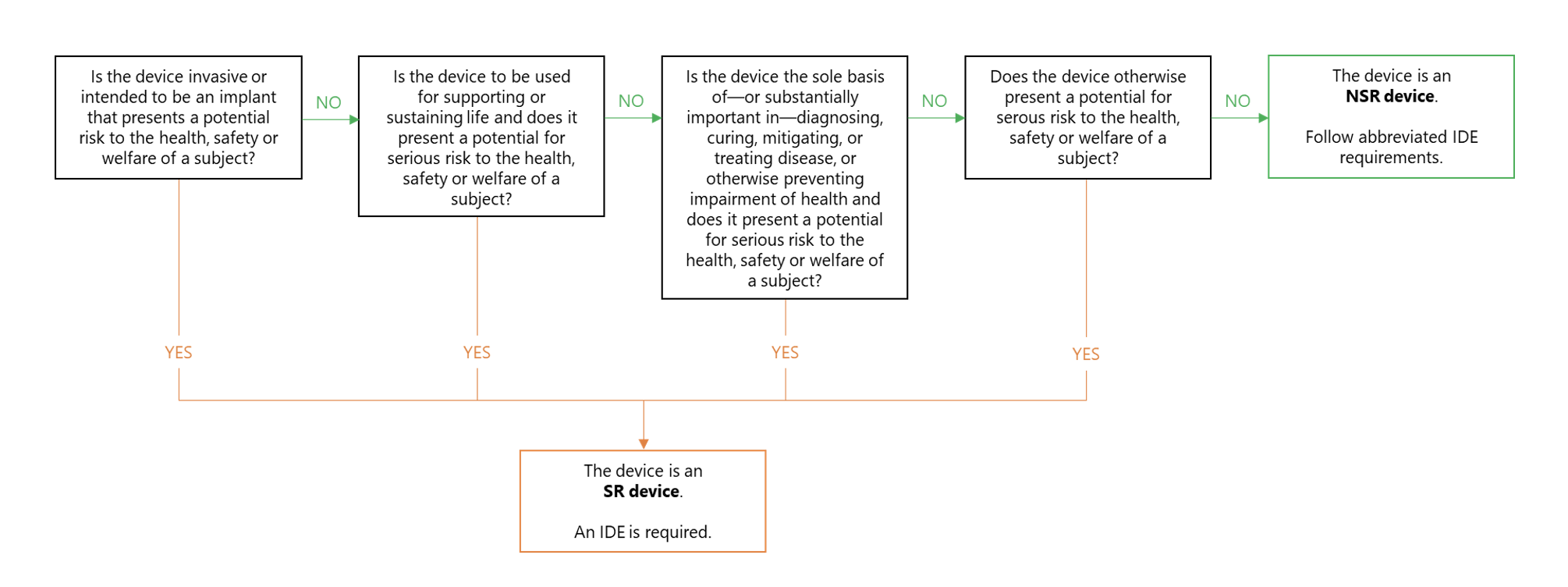 Figure 1. Distinguishing between SR and NSR device studies
Figure 1. Distinguishing between SR and NSR device studies
Situations where an IDE may not be required
Under section §812.2(c) of the IDE regulations, clinical studies are exempt from the IDE requirement if testing of the IVD meets all four criteria below:[2]
1. Is noninvasive:
A noninvasive device is one that does not penetrate or pierce the skin or mucous membranes, the ocular cavity or the urethra. Further, it is one that does not enter:
- The ear beyond the external auditory canal
- The nose beyond the nares
- The mouth beyond the pharynx
- The anal canal beyond the rectum
- The vagina beyond the cervical os
An IVD that requires blood sampling involving simple venipuncture is considered noninvasive, as is the use of surplus body fluids or tissues from samples taken for non-investigational purposes.[3]
2. Does not require an invasive sampling procedure that presents a significant risk
To determine whether an invasive sampling technique presents a serious risk, the FDA recommends that developers assess the nature of the harm resulting from the sampling procedure. Techniques that require a biopsy of a major organ, use of general anesthesia, or placement of a blood access line into an artery or large vein are considered significant risks.[3]
3. Does not by design or intention introduce energy into a subject
4. Is not used as a diagnostic procedure without confirmation by another medically established diagnostic product or procedure
To be exempt, the study must use another cleared or approved IVD or other method as the basis for decision-making. Moreover, test results from the exempt IVD investigation should not influence treatment or clinical management decisions until a medically established product or procedure has established the diagnosis.[3]
In addition to the criteria above, an IDE is generally not required if the study is not for any indication outside of the device’s existing label (on-label use) or if the study is not intended to support any significant change in labeling. Studies of custom devices—those made for specific individuals—are also exempt, as are studies of consumer preference, device modification, or comparison of marketed devices so long as there is no evaluation, safety, or effectiveness and no additional risk.
Regulatory requirements for exempt studies
IVD clinical studies exempt from the IDE regulations are still required to undergo institutional review board (IRB) review and approval. They are also required to obtain informed consent from study participants.
In addition, studies that are exempt from IDE regulations must still comply with labeling requirements under 21 CFR 809.10(c)(2). These requirements mandate that the device be labeled with one of two statements, as applicable:2
- “For Research Use Only. Not for use in diagnostic procedures.”
- “For Investigational Use Only. The performance characteristics of this product have not been established.”
Clarifying whether an IVD study is exempt
Determining whether an IVD clinical study is exempt under the IDE regulations is not always straightforward. Early engagement with the US Food and Drug Administration (FDA) ensures regulatory compliance and avoids incorrect assumptions or assessments about device risk and IDE exemption. At Premier Consulting, a division of Premier Research, our experienced experts can help you navigate the requirements. To learn more about IDEs, read Conducting Clinical Studies Under an Investigational Device Exception.
Resources
[1] US Food and Drug Administration. Investigational Device Exemption. Available at https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/investigational-device-exemption-ide.
[2] US Food and Drug Administration. FAQs about Investigational Device Exemption. Available at https://www.fda.gov/medical-devices/investigational-device-exemption-ide/faqs-about-investigational-device-exemption.
[3] US Food and Drug Administration. Guidance for Industry and FDA Staff: In Vitro Diagnostic (IVD) Device Studies – Frequently Asked Questions, June 25, 2010. Available at https://www.fda.gov/media/71075/download.