Evaluating Impurities in New Drugs to Prevent Delays in Development
During the development of new small molecule drug products, developers must conduct impurity and degradant evaluation at several points in the program and to varying degrees. These evaluations include the active pharmaceutical ingredient (API), also known as the drug substance, and the drug product (formulated product). Global regulatory agencies, including the U.S. Food and Drug Administration (FDA), expect identification, reporting, and qualification of these compounds according to three key guidance documents established by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
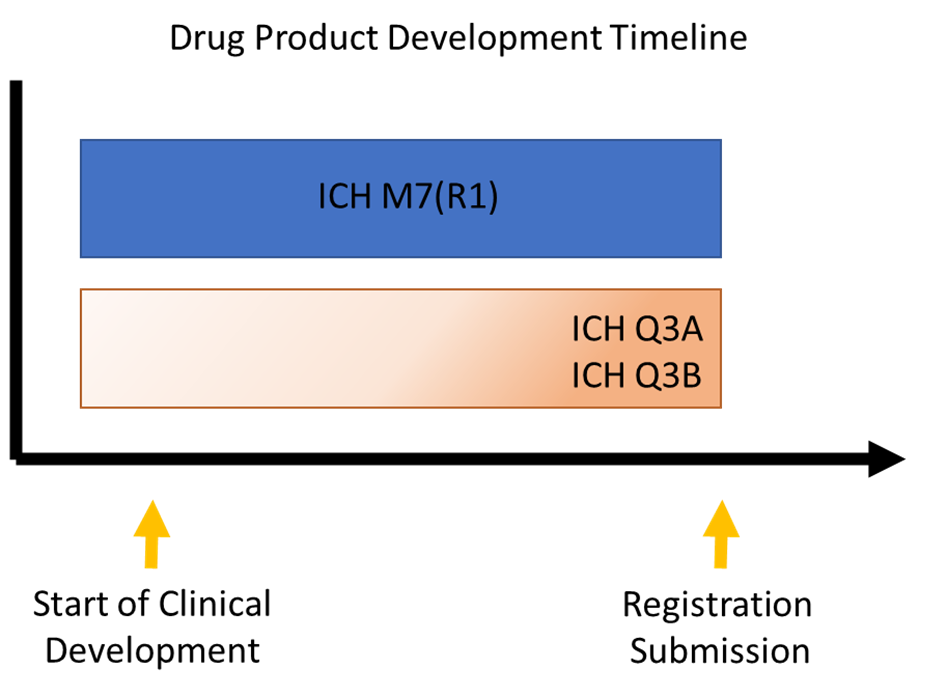
These ICH guidance documents apply at different stages of development and address specific concerns regarding certain types of impurities or degradants. Understanding the nuances and timing of when and how the guidance documents are implemented, either formally or informally, is critical to successful product development and avoiding obstacles that can delay or derail a program.
Impurities in new drug substances and new drug products
ICH Q3A(R2) addresses impurities in the drug substance, and ICH Q3B(R2) addresses degradants in the drug product. (Degradants are impurities arising from the degradation of the drug substance or a reaction with an excipient and/or the immediate container.) Formally, both of these documents apply solely to marketing approval; informally, however, they are often used as general guidelines during clinical development to avoid obstacles that could result in extensive process changes, product reformulation, or safety qualification efforts.
ICH Q3A and ICH Q3B outline expectations for reporting, identifying, and qualifying (i.e., evaluating the safety of) impurities and degradants. Table 1 compares key points from both guidances.
Table 1: Comparison ICH Q3A and ICH Q3B Guidance Documents
| Guidance Sections | ICH Q3A | ICH Q3B |
| Scope | In-Scope:
Out-of-Scope:
|
In- Scope:
Out-of-Scope:
|
| General Assessments | Identified and potential organic and inorganic impurities | Identified degradants arising from the degradation of the drug substance or the interaction of the drug substance with excipients and/or the immediate container closure system |
| Analytical Procedures | Validated methods | |
| Reporting, Identification, and Qualifying Thresholds |
|
|
| Qualification | Similar decision tree for qualification – may require separate in vitro and in vivo toxicology studies | |
DP = Drug Product; DS = Drug Substance
*Impurities have different reporting, identification, and qualification thresholds. Low levels of impurities need to be reported. As they increase in amount, then they may need to be identified. As they increase further in amount, then they may need to be qualified.
Impurities or degradants that exceed qualification thresholds and have not been adequately tested in Good Laboratory Practices (GLPs) toxicology studies are often encountered during development. Ideally, test materials used in the GLP toxicology studies should contain impurity and degradant levels that are equal to or higher than the expected release specifications for approval. These studies can be used to qualify the safety of the impurities and degradants as a group, so that qualification studies for each impurity or degradant are not needed. Sometimes a degradant presents unexpectedly after GLP toxicology studies are completed or are underway. Qualifying such degradant(s) does not necessarily require testing, as it may be possible to leverage existing literature and/or run in silico models to assess the safety of the degradant and make a safety justification.
Assessment and control of DNA reactive impurities
Another important guidance document related to impurities and degradants in new small molecule drugs is ICH M7(R1). ICH M7 provides a practical framework for identifying, categorizing, qualifying, and controlling mutagenic impurities and degradants to limit potential carcinogenic risk. What sets this guidance apart from ICH Q3A and ICH Q3B is that it applies to all stages of clinical development, not just to approval. In addition, this guidance focuses on DNA reactive substances that could potentially cause cancer. An associated Q&A document provides additional clarity on interpreting and implementing specific items in the guidance.
 Per the guidance, all identified impurities and degradants should be evaluated for mutagenicity, unless their concentrations are below the Threshold of Toxicological Concern (TTC). The TTC varies based on the clinical treatment duration and on whether multiple mutagenic impurities are present.
Per the guidance, all identified impurities and degradants should be evaluated for mutagenicity, unless their concentrations are below the Threshold of Toxicological Concern (TTC). The TTC varies based on the clinical treatment duration and on whether multiple mutagenic impurities are present.
Based on the chemistry, developers may also identify and should consider impurities that could potentially form. Review of the published literature and databases can identify available mutagenicity and carcinogenicity data. ICH M7 also provides limits, called Permissible Daily Exposures (PDEs), for a select set of mutagenic impurities.
For compounds without existing mutagenicity data or PDEs, a quantitative structure-activity relationship (QSAR) assessment is conducted to determine the mutagenic potential and recommended follow-up actions. These may include conducting a bacterial mutagenicity assay to rule out any QSAR structure-based concerns. Two complementary and validated QSAR models are required: an expert rule-based model and a statistical-based model.
Impurities and degradants in 505(b)(2) products
Products utilizing the 505(b)(2) pathway are not exempt from assessments of impurities and degradants. In fact, due to the oftentimes accelerated 505(b)(2) development process, it is even more important to be aware of potential and identified issues and ensure that the available data is sufficient to qualify any impurities or degradants, if needed. Interestingly, if impurities or degradants in the new drug product are present at levels equal to or lower than those in representative batches of the approved product (listed drug), they can be considered qualified.
Takeaway
It is important to be aware of any impurities or degradants identified in the process of drug development so that appropriate actions can be taken to ensure continued progress of the program.
Premier Consulting’s CMC and toxicology experts can help you to avoid or address the obstacles that can arise due to impurities and degradants. With extensive in-house experience in regulatory pathways for markets across the globe, we are able to support quality by design (QbD)-based pharmaceutical development, impurity qualification, and QSAR assessments. Contact us to learn more today!
Authors:
Madelyn Huang, PhD, DABT
Toxicologist
William Salminen, PhD, DABT, PMP
Vice President, Nonclinical Safety and Toxicology
Olu Aloba, PhD
Vice President, CMC Services
References
- ICH M7 (R1). 2017. ICH M7 (R1) Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk – Scientific guideline. https://www.ema.europa.eu/en/ich-m7-assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential
- ICH M7 (R2)- Q&A. 2022. ICH M7(R2) Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk- Questions and Answers. https://database.ich.org/sites/default/files/M7R2_QAs_Step3_2022_0517-Error%20Corrected.pdf
- ICH Q3A (R2). 2006. ICH Q3A (R2) Impurities in new drug substances – Scientific guideline. https://www.ema.europa.eu/en/ich-q3a-r2-impurities-new-drug-substances-scientific-guideline
- ICH Q3B (R2). 2006. ICH Q3B (R2) Impurities in new drug products – Scientific guideline. https://www.ema.europa.eu/en/ich-q3b-r2-impurities-new-drug-products-scientific-guideline