Do Your Biologics Assays Tell the Whole Story?
Adherence to cGMP regulations assures the identity, potency, purity and quality of biologics or advanced therapeutics and helps ensure adequate control of manufacturing operations. A primary way to demonstrate these attributes for a biologic product under development is through assays.
Stages of Assay Development
Design
This stage answers the question, “Does the assay give the desired output and behave as expected?” It also typically involves evaluation of many analytical and microbiological techniques. The assay needs to be “fit for purpose” and sufficiently developed to meet meaningful acceptance criteria. A draft analytical method of sufficient detail to enable a trained analyst to perform the assay with the expected outcome should be available by the end of the design phase.
Qualification
The assay candidate from the design stage is further developed and the design is confirmed as reproducible and suitable for the specified purpose. If the assay cannot be qualified, development must return to the design stage to evaluate alternative techniques. During assay qualification, changes may be made to address any discrepancy found and the assay modified and approved for use before testing in a GMP environment.
Validation
The assay is tested against predefined acceptance criteria to verify that its performance characteristics are suitable, robust, and reproducible for the intended application. Validation is not a one-time event; post validation, assay performance should be monitored to ensure the assay remains in a state of control. Examples of characteristics to monitor include Out of Specification (OOS)/Out of Trend (OOT) frequency, Reference Standard trending, system suitability trends and artifacts, “noise”, or anomalies.
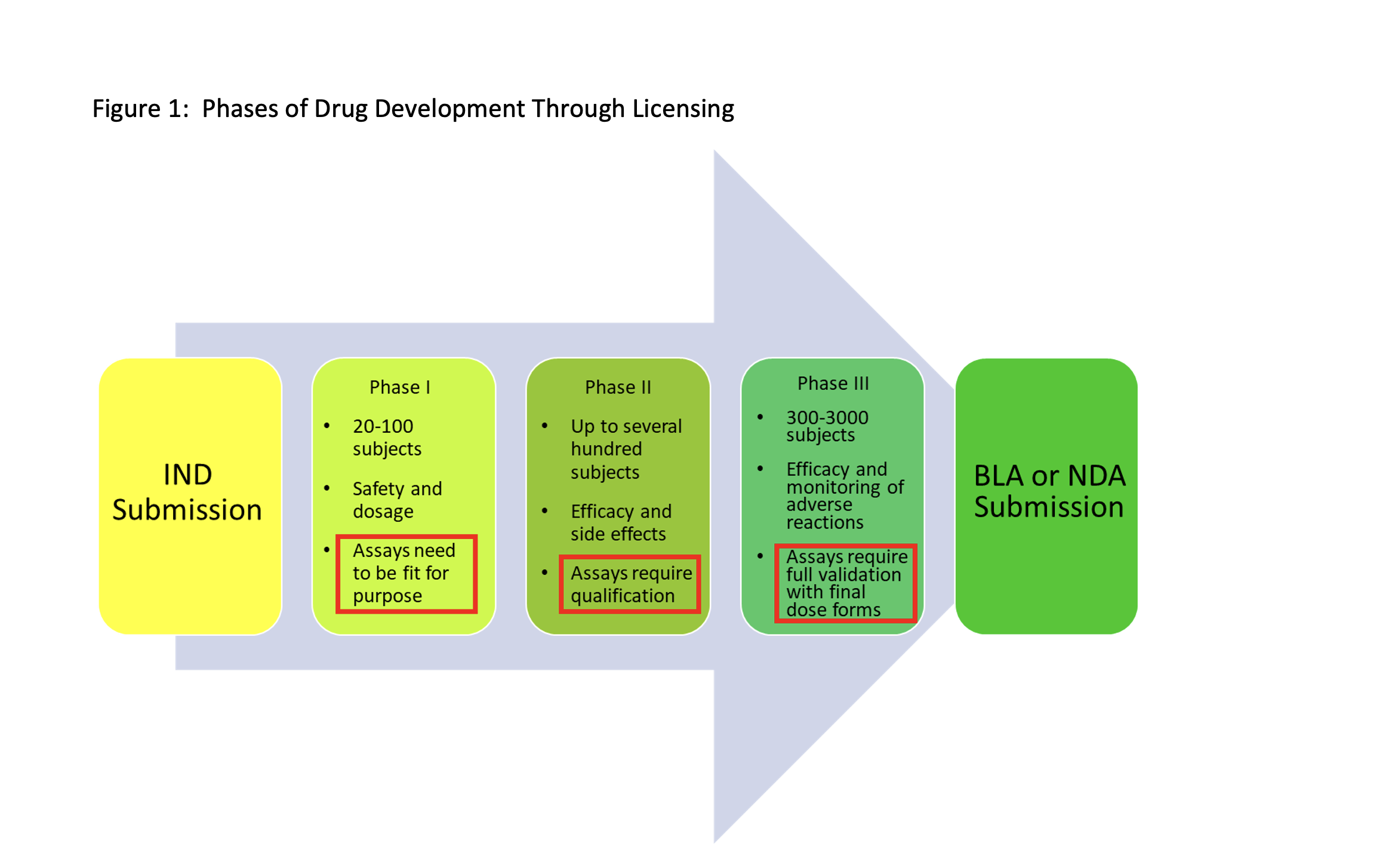
Phase Appropriate Assay Development
A phase appropriate approach for assay development is warranted especially with the significant development costs of novel large molecule or advanced therapeutic drugs. The “standard path” from IND through BLA is detailed in Figure 1. During the development process, different iterations of assays may be required to determine the identity, potency, purity, and quality of biologics.
- For Phase I studies, the requirement is that the assay is suitable for its intended use. Therefore, the assay should be demonstrated as specific, accurate, and repeatable. Acceptance criteria can be relatively broad but will need to be refined as drug substance or drug product development advances. [Assay Design/Qualification, as appropriate]
- For Phase II, the assays will require qualification and, if appropriate, validation. [Assay Qualification or Validation]
- For Phase III, it is recommended that the assays are fully validated (or revalidated if applicable), preferably using the final to-be-marketed product formulation. Full validation of analytical procedures will be required at BLA submission. [Assay Validation].
- Compendial assays may require qualification or verification at any of the phases, as needed.
Content Assays
Content assays (based on mass or quantity) do not always tell the entire story. For example, an assay for a protein-based drug product can be as simple as size exclusion chromatography – HPLC (SEC-HPLC) to enable quantitation by peak area. The chromatograms can also give other insights into the aggregation state of the drug substance or product. However, a chromatogram showing the protein as mostly monomeric does not provide information about its biological effect, so biological activity assays are required to define the potency of the drug substance or product.
Regulatory Health Authorities
Testing for activity/potency is expected for all biological medicinal products, including Advanced Therapy Medicinal Products (ATMPs). Many regulatory jurisdictions have this as a legal requirement. Example FDA and EMA requirements are as follows:
European Medicines Agency (EMA)
- EMA/CAT/852602/2018: Guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials (2019).
- EMA/CHMP/BWP/271475/2006 rev.1: Guideline on potency testing of cell-based immunotherapy medicinal products for the treatment of cancer (2016).
US Food and Drug Administration (FDA)
- 21 CFR 600.3(s): Potency is interpreted to mean the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result.
- 21 CFR 610.10: Tests for potency shall consist of either in vitro or in vivo tests, or both, which have been specifically designed for each product, so as to indicate its potency in a manner adequate to satisfy the interpretation of potency given by the definition in § 600.3(s) of this chapter.
The above FDA regulations are further codified in guidances to assist sponsors of developmental biologics. For example, draft guidance ”Potency Assay Considerations for Monoclonal Antibodies and Other Therapeutic Proteins Targeting Viral Pathogens” (March 2023) provides recommendations for developing and implementing potency assays to ensure each lot of monoclonal antibodies (mAbs) or therapeutic proteins targeting viral pathogens is produced consistently with the potency necessary to achieve efficacy and that the potency is maintained through the shelf life of the product.
The draft guidance provides the following general recommendations for all assay types:
- Relevant positive and negative controls should be included as part of the potency assay to ensure there are no matrix effects.
- For mAb cocktails, release testing methods should include an identity method for each mAb and a quantitative method for the ratio of the individual mAbs. Ensure the ratio is consistent from lot to lot.
- For assay relevance, ensure that the virus isolate, or viral surface (glyco)protein(s) used reflects common isolates prevalent in the U.S.
- For wild-type virus, pseudotyped virus, or virus-like particles, an appropriately qualified master cell bank of producer cells should be used.
- Record differences in methods used for release and stability testing as compared to those used to initially characterize the potency during earlier development.
Cell-based potency assays that determine the relative potency of a product by measuring biological activity based on the product’s mode of action are especially relevant to mAbs.
Types and example uses of such assays are shown in Table 1.
| Table 1: Types of Cell-based Potency Assays and Example Uses | |
| Assay Type | Sample Type or Use Condition |
| Binding and/or Competitive Assays | anti-viral monoclonal antibodies |
| Cell Signaling Assays | immune checkpoint PD-1/PD-L1 blockade assays, ß-adrenergic signaling in heart failure |
| Cell Migration Assays | cell metastasis in oncology |
| Cell Proliferation or Inhibition Assays | proliferation for regenerative medicine, heart or liver disease; inhibition of tumor growth for oncology |
Recommendations specific to assays for mAbs and virus-targeting proteins are shown in Table 2.
| Table 2: Recommendations by Types of Assays for Monoclonal Antibodies or Proteins Targeting Viral Pathogens | |
| Assay Type | Recommendation |
| Binding Assays | Develop as inhibition assays (e.g. inhibition SPR/ELISA) instead of direct binding assays. |
| Viral Neutralization Assays | Confirm mechanism of action and establish assay using:
· Blocking of fusion of wild type or lab-adapted virus, pseudotyped virus or virus-like particles · Utilize cells expressing viral proteins to assess viral surface protein-mediated cell-cell fusion |
| Constant Fragment (Fc)-effector Function Assays | To demonstrate Fc-mediated effector functions, assess:
· Complement activity and activities mediated through binding to Fcγ receptors, e.g. antibody-dependent cellular cytotoxicity. · Fc glycosylation relevant to the mechanism of action. |
While products for targeting viral pathogens will work on known mechanisms and assay readouts (e.g. viral infectivity), there are often situations where a cell-based assay utilizing surrogate markers is required. Surrogate biomarkers such as cytokines, interleukins, growth factors, etc. are often measured as a direct response to application of the therapeutic agent to a suitable substrate. Such surrogate assays must be scientifically sound and justified.
Given the complexity and expense of these assays, early interactions are recommended to obtain FDA concurrence on the suitability of any proposed assays. These interactions may be in the form of INTERACT, pre-IND/Type B, Type C, or Type D meetings.
Premier Consulting’s CMC experts can assist with phase-appropriate assay design, validation, and documentation, as well as interactions with the FDA and other regulatory agencies. Contact us today.