De-risking Drug Development: High-level Reasoning and Technical Methodology for Evaluating a Program’s Risk
From a distance, 505(b)(2) product development can seem very straightforward for products that have been on the market or in clinical use for long periods of time. However, these types of products can often present the greatest challenges when trying to modify or improve performance. This is particularly true for products with known safety risks or narrow therapeutic windows, among other complicating factors. These products are used because the risk/benefit calculus still favors the benefit for patients with a medical need, but sponsors have to be mindful of reformulating these products to avoid unintentionally tipping the risk/benefit equation in the wrong direction. Even the “simplest” drug development program has the potential to go far awry.
Based on our experience developing products through the 505(b)(2) approval pathway, Premier Consulting is constantly working to identify new ways to identify and avoid potential pitfalls as early as possible in a development program. One proven method is to leverage existing data.
When we evaluate known factors for a drug product under development, it can mean using early-stage pharmacokinetics (PK) data to simulate known risk factors or patient behaviors to identify potential safety hazards, or to extrapolate efficacy in new populations or under different conditions for a new product. By using data collected early in a development program to evaluate potential risks or benefits of a formulation, sponsors can take action to mitigate potential risks before investing time and money in a product that may not achieve their ultimate goals.
Common Risks in Drug Development with Examples
Reformulation Pitfalls
Product reformulation can often produce new and unexpected changes in systemic exposure to the active moiety, which can impact the safety or efficacy of a product even if it meets the standard bioequivalence criteria when compared to a Listed Drug (LD). Sponsors developing new formulations of existing drugs may think they are in the clear if their product meets the standard single-dose bioequivalence criteria, but experience shows the FDA does not always agree. This is particularly true for drugs which have narrow therapeutic ranges or those with specific dose- or exposure-related safety concerns, and may also apply to extended- or modified-release products. Premier Consulting has developed tools to evaluate the potential for product-specific safety and efficacy concerns using only PK data, which can help sponsors identify and mitigate risk to their development programs in the earliest stages.
Safety Concerns
It is of particular interest to ensure that patients requiring rapid onset of action, such as those being treated for acute pain, ADHD, or sleep disorders, receive minimum effective exposure relatively rapidly. If a reformulated product produces a delayed response for any reason or under any specific conditions (for example, following a high-fat meal, immediate release / extended release (IR/ER) ratio of the new formulation, etc.), the risk of patients re-dosing to meet their therapeutic need is increased and may result in overdose when the drug substance is eventually absorbed into the systemic circulation. This can result in a significant safety risk for products with known exposure-related adverse events. A classic example of this kind of safety risk would be opioid analgesics, which are known to cause respiratory depression at high doses.
Efficacy Concerns
Alternatively, the same tools can be used to evaluate the potential efficacy of a product in relation to a listed drug (LD). For example, if a product is intended to act locally, but produces a predictable pattern of measurable systemic exposure, it may be possible to demonstrate therapeutic equivalence with only PK data. In some cases, by evaluating PK data, it may be possible to eliminate the need for time-consuming and costly Phase 3 trials entirely.
Example Analysis
As an example of a tool to mitigate risk associated with novel formulations, Premier Consulting evaluated PK data from recently developed and approved abuse-deterrent IR opioid products. A common theme with these kinds of products is that the goal of deterring abuse requires the addition of unique dosage forms or excipients that may impact the release or absorption of the drug substance under specific conditions.
These products often meet the requirements for demonstration of bioequivalence compared with the LD (Cmax and AUCs), but show a delay in Tmax under standard fed conditions as illustrated in Figure 1. Because of the risk of patients re-dosing, the FDA has scrutinized the effect of food on the bioavailability of the active moiety for these products. Because of our interest in 505(b)(2), Premier Consulting did some analysis of our own and presented the results at the 2016 AAPS annual meeting.
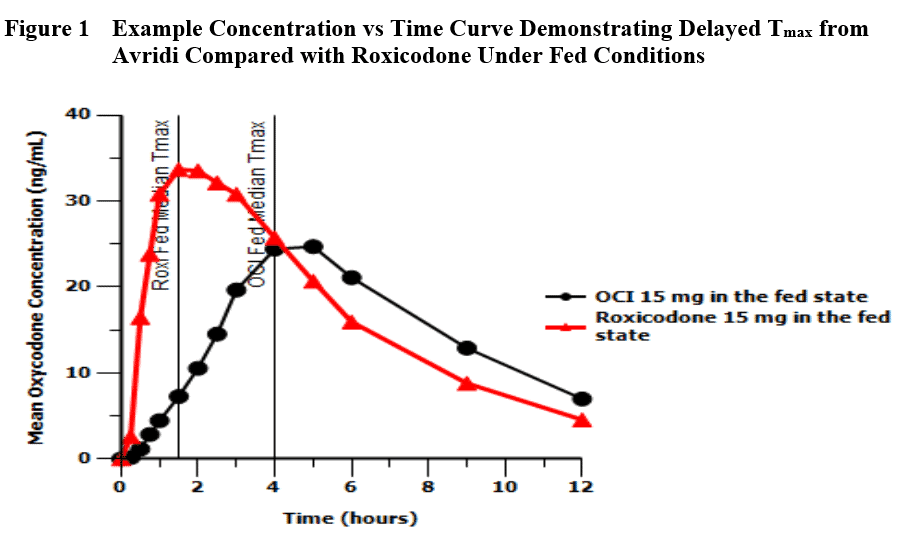
From a regulatory perspective, the issue is that the current BE guidance does not consider delayed Tmax when evaluating bioequivalence (BE) for IR opioids, nor do well-controlled Phase 3 trials address the potential safety risk from delayed Tmax under fed conditions due to tight controls over timing of dosage administration.
Premier Consulting proposes that evaluation of the partial extent of exposure (pAUC) under fed conditions can help to identify products at risk for potential safety concerns during later stages of development or upon FDA review. To support this proposal, we utilized oxycodone (Oxaydo [NDA 202080] and Avridi [NDA 206830]) PK data as an example supporting the rationale of adding pAUC to current BE evaluation criteria, through population PK modeling and simulation.
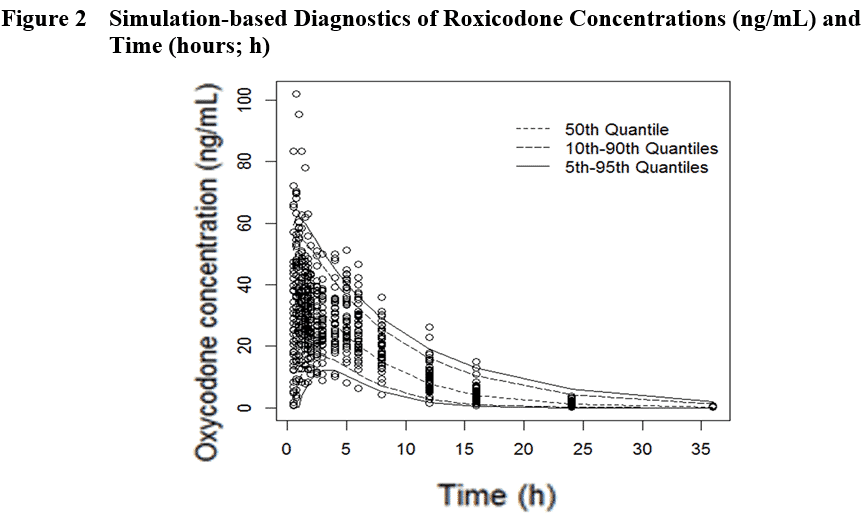
Using published PK data, the Premier Consulting team developed covariate population PK models (Figure 2) and ran simulations of 100 virtual 2-way crossover BE trials (32 subjects/trial) comparing the hypothetical Test versus IR Reference (Roxicodone under Fed conditions).
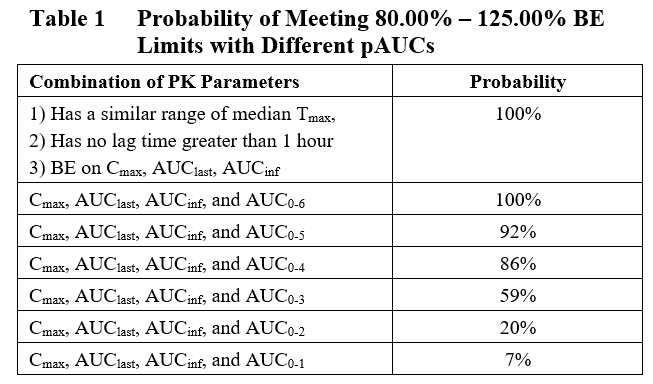
Based on the results of these simulations, the probability of several pAUC intervals were evaluated for the ability to predict the potential for a significant delay in Tmax for the Test product compared with the Reference (Table 1). The results of this analysis show that pAUCs £ 4 hours are able to differentiate reformulated products that display potential safety risk associated with delayed Tmax from the reference product. However, pAUCs > 4 hours show limited to no ability to differentiate the two products or identify potential safety risk.
Therefore, Premier Consulting proposes that pAUC0-4 provides a useful tool to screen reformulated IR opioid products for potential effects of food on bioavailability. By identifying this early in the development program, sponsors can re-evaluate the cost of development vs reformulation to determine the best path forward before investing significant resources in a potentially failing product.
The Key to Utilizing Existing Data
Because of our history of regular interactions with the FDA, Premier Consulting is aware of key issues of safety and efficacy associated with drug products in development. Reformulated opioid products provide an example of what can be done to identify potential safety hazards from a product early in development using only PK data. Similar examples exist where understanding of PK data analysis can significantly impact the development requirements for approval.
By understanding how to leverage existing data to evaluate new products, it is possible to identify and mitigate risks early in a development program and avoid unnecessary investment in products that may not meet the sponsor’s goals. The 505(b)(2) development pathway presents unique challenges and opportunities, and Premier Consulting has the know-how to get sponsors to the finish line with greater confidence in the success of their product.
For more information on Premier Consulting’s cross-functional team approach to drug development, including PK, toxicology, and CMC specialists, contact us.