ANDA Suitability Petition vs 505(b)(2)
I was honored to be invited to speak at the FDA-OCRA 12th Annual Educational Conference in Irvine California on June 10, 2009. I was asked to discuss and compare the 505(b)(2) and ANDA Suitability Petition. I thought I should share this topic with the readers of this blog.
Why make the comparison between an ANDA Suitability Petition and a 505(b)(2)? Because both allow for changes from a reference listed drug (RLD).
I assume we all know that an ANDA product is the same as the RLD and is governed by 505j. Yet, the same Hatch-Waxman amendments in 1984 provided for a suitability petition that allows the following differences in an ANDA product:
- A different active ingredient in a combination product in which the other active ingredients match those of the RLD
- A different route of administration
- A different dosage form
- A different strength
Hold it! Aren’t these differences spelled out for 505(b)(2) differences too?
The bottom line is that an ANDA can contain only those differences that do not need clinical evidence for efficacy and safety. It turns out that most changes need clinical studies so there are very few approved ANDA suitability petitions.
An ANDA suitability petition is a petition (request) to FDA to permit the filing of an ANDA for a drug that differs from the RLD. Thus, you must have an approved suitability petition in order to file an ANDA with the proposed differences. The law doesn’t do a good job at laying down the process for these petitions, but FDA handles them like Citizen Petitions. That is, the process includes:
- File petition
- Public Docket
- Public Input
- FDA OGD/CDER Review
- Decision published
- If Petition is approved, ANDA may be filed
Thus, the ANDA suitability petition is a very public process. Many pharma companies use consultants or their legal firms to file in order to avoid publicity. Note that once a suitability petition is approved, anyone can use the approval to file for the same change.
The law provides for a 90-day review but that never happens. Because OGD needs to have CDER’s input, and CDER’s priority is the PDUFA drugs, reviews can take a long time (we are going on 3 years for a suitability petition for one of our clients).
The petition itself is rather simple. It contains the requested change, the reason the change should be accepted or denied (like any governmental petition, both favorable and unfavorable information must be included) and the proposed labeling. It is not the ANDA and FDA’s approval of a suitability petition doesn’t mean it will approve the ANDA when filed (meaning it could change it’s decision).
The listing of petitions is on the FDA website. An excerpt will help us illustrate the grounds for approval and denial:
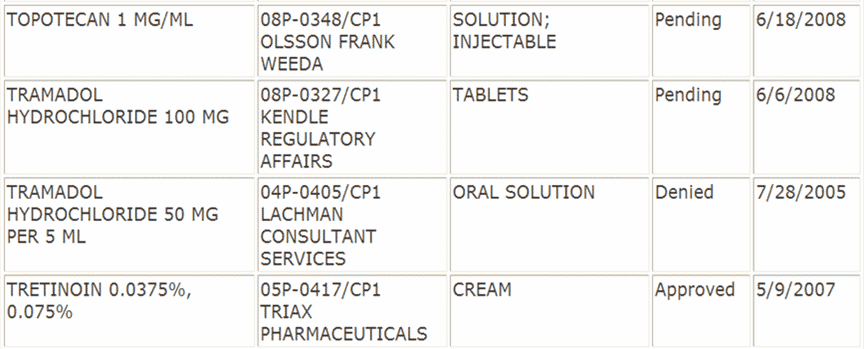
The excerpt above shows petitions that are pending, approved and denied. The process is such that a petition is approved if it is not denied, so let’s look at the reasons for denial:
- Changes to an API in a single ingredient product
- The petition does not contain information to show that the different active ingredient of the drug product is of the same pharmacological or therapeutic class as the ingredient of the RLD
- The different active ingredient is not an active ingredient in a listed drug
- The remaining active ingredients are not identical to those of the listed combination drug
- Any of the proposed changes from the listed drug would jeopardize the safe or effective use of the product so as to necessitate significant labeling changes to address the newly introduced safety or effectiveness problem
- FDA has determined that the RLD has been withdrawn from sale for safety or effectiveness reasons
As I wrote above, basically the reason for denial is that the proposed change raises efficiacy or safety issues that must be addressed via clinical studies – that is, the changes must be submitted and reviewed under 505(b)(2).
Let’s look at the examples above.
Approval: Triax Pharmaceuticals submitted a suitability petition as follows:
- RLD: Retin-A (Tretinoin) Cream, 0.025%, 0.05%, and 0.1 %, Johnson & Johnson
- Proposed Change: two intermediate strengths 0.0375% and 0 .075%
- Basis for Approval: The Agency has determined that sameness of therapeutic effect for these two interim strength Tretinoin Cream products can be demonstrated using comparative bioavailability data
Denial: Lachman Consultant Services submitted a suitability petition as follows:
- RLD: UltramB (Tramadol Hydrochloride) Tablets, 50 mg, Ortho-McNeil Pharm, Inc
- Proposed change: Tablets to Oral Solution
- Basis for denial: (a) PREA. UltramB is not labeled for pediatric use; clinical studies would be required and (b) clinical trials needed for oral solution due to uncertain pk/pd relationship
Generally speaking, changes that are approvable are dose changes. While at Duramed, we obtained approval for several products, for example, where the RLD had scored tablets or intermediate dosage strengths where the RLD labeled dose was 2 tablets or capsules.
ANDA suitability petitions were quite popular in the late 1980’s and 1990’s because FDA determined that many changes didn’t require clinical studies. This changed with the advent of pediatric initiatives. In 1999 Congress passed what is known as the Pediatric Rule. The applicable part is:
“… each application for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration shall contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations.” 21 CFR 314.55(a)
As if there was any question that this applied to ANDA suitability petitions, FDA covered it:
“… if a petition is submitted for a change that would require a pediatric study under this rule, the petition may be denied.” 63 Fed. Reg. at 66641 (FDA response to Comment 10)
The Pediatric Research Equity Act (PREA) of 2003 and renewed and extended by Title IV of 2007 FDAAA, stipulated that an assessment was required for all applications submitted after April 1999 containing a:
- new active ingredient
- new indication
- new dosage form
- new dosing regimen, or
- new route of administration
Indeed, PREA caused FDA in early 2007 to rescind approval of about (there were errors so the exact number is changing) 128 suitability petitions:
“FDA had approved these suitability petitions to permit ANDAs to be submitted for drugs that had a different active ingredient, dosage form, or route of administration than their RLDs. However, these approval decisions are being withdrawn because ANDAs were never submitted and PREA requires that all applications submitted on or after April 1, 1999, for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration contain an assessment of the safety and effectiveness of the drug for the claimed indications in relevant pediatric subpopulations.” 72 FR 8184 23Feb2007 (edited for brevity)
The applicable regulations are at 21 CFR 314.93.