Improving NDA Approval Odds for New Dosage Forms of Approved Products
There are numerous reasons why a sponsor may wish to market a new dosage form of an approved product. Aside from the obvious financial benefits to the sponsor, providing a more convenient and/or faster-acting dosage form of a well-chosen drug provides significant benefits for patients.
Examples of new dosage forms include oral solutions and suspensions of drugs that were previously only approved as solid oral dosages forms. Liquids are more appropriate for some pediatric and geriatric populations and for patients with difficulty swallowing. Other products designed to enhance ease of swallowing include chewable or effervescent tablets, oral films, and orally disintegrating granules. Sponsors may wish to market both capsule and tablet forms of their products for patient convenience. Faster-acting oral dosage forms such as buccal lozenges or films are beneficial for indications requiring a rapid onset of action.
Why is Approval Necessary for Alternate Dosage Forms?
Approval of a new dosage form of a drug requires a new NDA. If the route of administration and indication are the same, comparative bioavailability studies are often needed to demonstrate equivalence to the approved product. If the dosage forms are not bioequivalent, additional data and/or justification will be required to demonstrate that the product is as efficacious and safe as the approved product.
If a sponsor is developing a product with a different route of administration or indication, additional studies are usually required to demonstrate the safety and efficacy of the product specific to the new route of administration or indication. This may involve clinical studies assessing local and/or systemic exposure and additional nonclinical studies.
Sometimes, the regulatory standards change after the approval of the listed drug (the approved drug product to which new dosage forms are compared to demonstrate bioequivalence). In such cases, approval of a new dosage form may require additional studies to meet the current standards.
Which Approval Pathways are Appropriate for New Dosage Forms?
The 505(b)(2) regulatory pathway is typically utilized for approval of products that represent new dosage forms of approved drugs. This is because these products do not qualify as generics (505(j) pathway), and at the same time do not require a 505(b)(1) application containing complete evidence of efficacy and safety. The 505(b)(2) pathway allows reliance on data from a listed drug or from publicly available literature.
The use of publicly available data in lieu of sponsor-conducted studies requires the establishment of a scientific bridge from the sponsor’s product to the data. Designing a bridging study to establish this bridge can be very nuanced. Seeking advice from a 505(b)(2) expert can prevent the need for additional bridging studies and can minimize the need for additional efficacy and safety studies.
Classification of a New Dosage Form
When a sponsor files an NDA, the FDA typically assigns an NDA classification code to the application. At the time of approval, a reassessment of the classification, relative to other approved products, is made. According to the Center for Drug Evaluation and Research’s Manual of Policies and Procedures 5018.2, a Type 3 NDA is for a New Dosage Form of an active ingredient that has been approved or marketed in the United States by the same or another applicant but in a different dosage form. The indication for the drug product does not need to be the same as that of the already-marketed drug product. Once a new dosage form has been approved for an active ingredient, subsequent applications for the same dosage form and active ingredient will be classified as Type 5 (New Formulation or Other Differences).
505(b)(2) Approval Statistics for New Dosage Forms
A review of Premier Consulting’s 505(b)(2) database finds that 246 NDAs were approved as New Dosage Forms between 2016 and 2021. Of these NDAs, oral tablets were the most frequent dosage form, followed by injectables, oral solutions and suspensions, and oral capsules.
Figure 1. 505(b)(2) NDAs with New Dosage Forms (2016-2021)
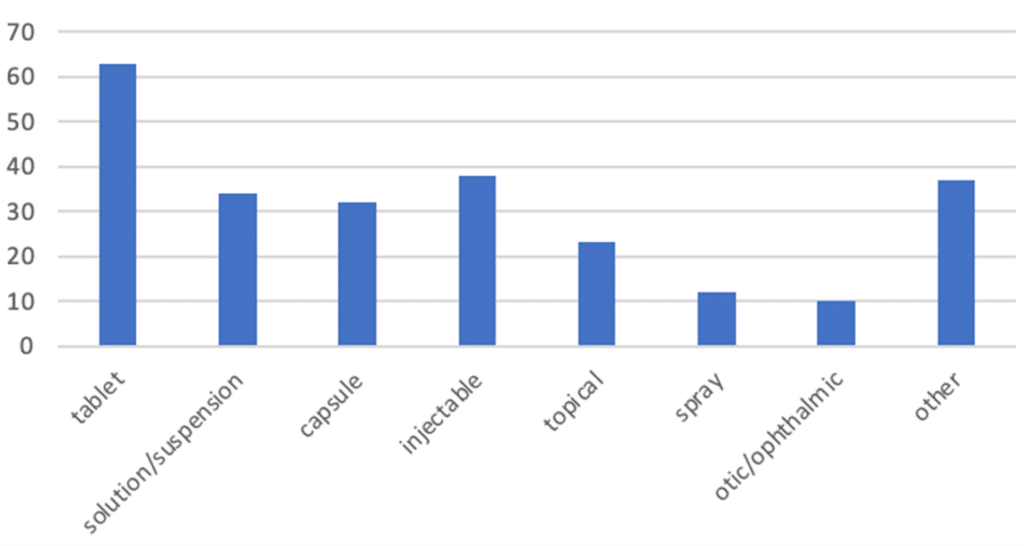
‘Oral solution’ category includes oral suspensions.
‘Topical’ category includes creams, cloths, lotions, ointments, shampoos, swabs, gels, and aerosol foams.
‘Spray’ category includes oral, nasal, topical, sublingual, and /inhalation products.
‘Otic/ophthalmic’ category includes injectables, solutions, solutions/drops and suspension/drops.
‘Other’ category includes aerosols (inhalation, nasal, sublingual), films (buccal and oral), solutions (inhalation, intramuscular, intravenous, oral, topical), implants (implantation, sinus), kits (inhalation, oral), powders (inhalation, intrapleural, oral), vaginal rings, chewable bars, oral granules, and patches.
Of the oral tablet, capsule, and solutions approved as new dosage forms, 44 are extended or delayed release. Twenty-one products have features to allow rapid absorption, such as sublingual or buccal delivery, or orally disintegrating tablets. Aside from these rapid-absorption products, a further 38 oral products offer alternatives to swallowing a tablet or capsule such as solutions, suspensions, and effervescent or chewable tablets.
Many of the new dosage form NDAs represent combination products. Thirty-two are fixed-dose drug combinations, and 23 are drug-device combinations.
What Studies are Needed to Get a New Dosage Form Approved?
In many cases, few if any clinical studies are required for approval of a new dosage form. For 33 of the products approved as new dosage forms, no clinical studies were required; seven were only nonclinical studies, and 26 were literature-based. These products were frequently injectables and oral tablets, and several were oral solutions. When approved via the 505(b)(2) pathway, development programs for these products may receive a biowaiver of the requirement to perform in vivo studies and rely on literature and approved products to demonstrate safety and efficacy.
For 35% of products, only Phase 1 (pharmacokinetic) studies were required for approval. Over-represented in this group are oral solutions and orally disintegrating tablets. In the remaining products, 55.3% required at least one Phase 2 or 3 study, or a human factors study for approval. These products were frequently oral extended- or delayed-release, topical, drug-device combination, inhalation, and ophthalmic products.
Nonclinical studies were required in 105 (42.7%) applications. In the remaining 141 (57.3%), nonclinical studies were not required for approval, but two programs had nonclinical post-marketing commitments. In one study, pharmacology and toxicology nonclinical studies were performed before NDA approval; carcinogenicity studies were deferred to post-approval.
In 10.6% of applications, no clinical or nonclinical studies were required for approval.
Review Time
The average time between submission and tentative/full approval was 595.8 days, with a median time of 384 days and a range of 105 to 3652 days. Interestingly, there was no correlation between dosage forms and review time or between applications with or without studies and review time. Typically, the quality and completeness of an application determines the review time.
Many of these costly review delays can be avoided by engaging a 505(b)(2) expert in designing the development strategy. Premier Consulting has helped numerous sponsors avoid review delays and refuse-to-file actions by performing due diligence on development programs and/or reviewing NDAs before submission to the FDA.
To learn more about getting a new dosage form approved via the 505(b)(2) pathway, or to get assistance with avoiding deficiencies in the development program or CMC data, contact us.
Authors:
Angela Drew, PhD
Director, Regulatory Strategy
Olu Aloba, PhD
Vice President, CMC Services
Vanessa Diniz Atayde, PhD
Senior Associate, Regulatory Affairs