In the News: Regulatory and Development Updates
Rare Disease Clinical Trials Most Often Terminated Due to Regulatory and Recruitment Issues
The difficulty associated with successfully completing rare disease clinical trials is well-known. Most people believe that recruitment is the primary reason because, by definition, the number of potential trial participants is extremely low. A recent analysis by GlobalData of over 730 rare disease trials terminated between 2016 and 2020 shows that approximately a quarter of the trials were terminated due to recruitment issues. However, the same analysis finds that a full 40% of the terminated trials were stopped due to regulatory issues.
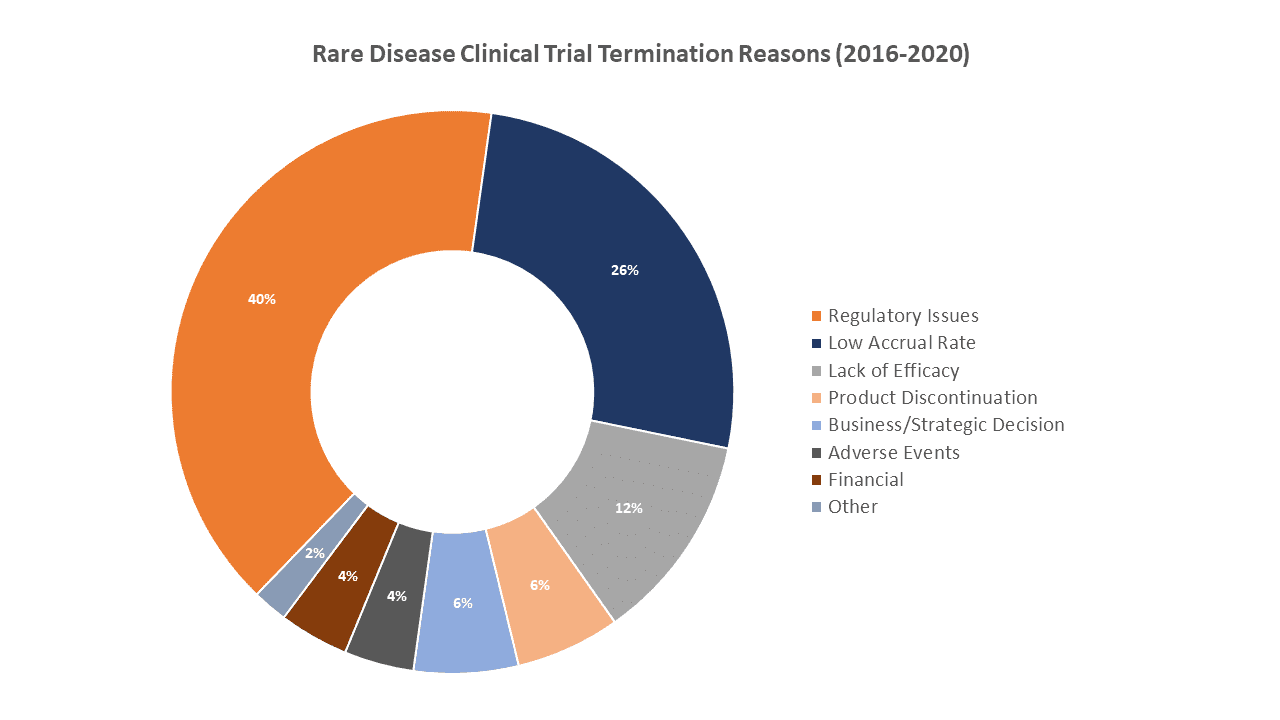
Source: GlobalData Pharma Intelligence Center
In the report, GlobalData suggests that virtual trials may be the solution to the low accrual issue. However, a broader solution is needed – the coupling of a robust regulatory strategy endorsed by the FDA with a customized clinical trial design that incorporates real-world evidence with targeted virtual (siteless) and remote trials.
FDA regulations do not distinguish between rare and common diseases, leading some drug developers to apply the tried-and-true development paradigm – standard Phase 1, 2, and 3 studies – to rare diseases. However, Premier Consulting’s extensive experience in rare and complex programs has shown that the FDA’s requirements for approval can often be met with alternative approaches.
For example, one alternative approach allows sponsors to fulfill the requirements for pivotal trials using publicly available information, including compilations of real-world evidence (RWE) to serve as a comparator in a single-arm clinical trial. A single-arm trial requires fewer patients and can thus be the answer to recruitment issues. This solution also overcomes the difficulty of recruiting patients to a placebo-controlled trial. To be successful, such trials require out-of-the-box thinking and experience with the FDA.
Approval of the Month: Aveo’s Fotivda for Refractory Advanced Renal Cell Carcinoma Granted Third-Line Position After Failed First-Line Studies
Patients 18 and older with advanced renal cell carcinoma (RCC) who have failed two or more prior systemic therapies will welcome the FDA’s March 2021 approval of Aveo Oncology’s Fotivda (tivozanib).
Fotivda was initially developed for first-line RCC. In the TIVO-1 trial, Fotivda-treated patients had a 2.8-month progression-free survival (PFS) improvement over patients treated with Bayer’s standard therapy Nexavar but lacked a significant overall survival (OS) benefit. The FDA’s May 2013 Oncologic Drugs Advisory Committee voted 13-1 that Fotivda did not demonstrate a favorable benefit-to-risk for the (first-line) treatment of advanced RCC. The FDA then rejected the NDA later in 2013.
This month’s approval was based on a single Phase 3 study (TIVO-3) of Fotivda versus Nexavar. In the primary endpoint of PFS, patients in the Fotivda arm experienced a median PFS of 5.6 months versus 3.9 months PFS for those patients in the Nexavar arm. In a bit of a disappointment, the OS endpoint was 16.4 versus 19.2 months for Fotivda and Nexavar, respectively. But on a brighter note, 18% of Fotivda patients achieved a pre-specified objective response rate (ORR), compared with an 8% ORR for the Nexavar patients.
What Comes After a Complete Response Letter?
Little can be more unpleasant for a sponsor during the drug approval process than an application rejection, typically in the form of a Complete Response Letter (CRL). A CRL states that the application is not approvable and lists the deficiencies the FDA expects the sponsor to address before resubmitting the application. The sponsor has the opportunity to meet with the FDA following the issuance of a CRL so that the sponsor and the FDA can come to an agreement regarding the specifics involved in adequately addressing the deficiencies.
While the sponsor can often successfully address the deficiencies and gain approval following resubmission, that is not always the case. A recent Federal Register notice offers an interesting primer on how the process can proceed following a CRL, but not in a particularly positive way. The notice, “Proposal To Refuse To Approve A New Drug Application for Sotagliflozin Oral Tablets . . .,” outlines the journey thus far of NDA 210934. The notice traces the issuance of a CRL and describes the attendant deficiencies in some detail. Also discussed are several attempts by the sponsor to use the FDA Formal Dispute Resolution process and the transfer of the NDA’s ownership to another company.
Clearly, there are some fundamental disagreements between the FDA and the sponsors of the application. Is all this back-and-forth required? The short answer is “Yes.” If a sponsor is convinced that it is in the right, it may wish to pursue a court action. However, to get to court, it must exhaust the available administrative remedies. The FDA, in turn, must follow its own procedures and satisfy the requirements of the Administrative Procedures Act. It can be a drawn-out, expensive, and frustrating process for all concerned. The notice does not make it completely clear what is at the root of the disagreements, but the best way to minimize the likelihood of finding yourself in such a situation is to (a) communicate early and often with the FDA and (b) put together the best application possible. Premier Consulting can help with both.
FDA Documents, Exclusivity Updates Offer Insights for Selecting Listed Drugs
Central to most NDAs filed through the 505(b)(2) pathway is reference to a Listed Drug (LD). As the name implies, an LD is an approved drug product listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, aka, the Orange Book. An NDA sponsor can rely on the FDA’s findings of safety and effectiveness for an LD by establishing a scientific bridge between its product and one or more relevant LDs.
FDA: BELVIQ Withdrawn Due to Safety or Efficacy, No Longer Eligible as 505(b)(2) LD
Recently, the FDA published a notice in the Federal Register declaring that BELVIQ (Lorcaserin Hydrochloride) was withdrawn from sale for reasons of safety or effectiveness. The notice is a useful overview of how the FDA’s review process works in such cases and gives the specifics for BELVIQ.
For a sponsor to rely on a withdrawn LD that is no longer marketed, there must have been a finding by the FDA that the LD was not withdrawn for reasons of safety or efficacy. The FDA may perform a review of the relevant data on its own initiative or in response to a Citizen’s Petition. Following the review, the FDA publishes a notice in the Federal Register outlining its determination. If the FDA determines that a product was withdrawn for reasons other than safety or effectiveness, the product is still a valid LD for the sponsor of a 505(b)(2) NDA.
Products Losing Exclusivity Offer Opportunities for 505(b)(2) Applications
Fierce Pharma recently posted the top 10 drugs losing U.S. exclusivity in 2021. Products losing exclusivity are often good candidates for LDs (see above) in NDA applications. The 505(b)(2) pathway often offers a faster, less expensive route to drug product approval. Selecting an appropriate LD is critical, and working with Premier Consulting can assure a successful program.
Author:
Ken Phelps
Strategic Advisor