Getting Cannabis-related Products Approved: the 505(b)(2) 4-20 Projects
Several states in the US have already passed laws that remove state restrictions on the medical use of marijuana and its derivatives, and more states are considering such action. The market for medical marijuana in the US is expected to surpass $20 billion by 2020. However, federal restrictions prevent the use of medical marijuana as only synthetic derivatives have been approved as therapeutics by the FDA. The difficulties in producing medical grade cannabis with ensured quality and efficacy are rapidly being overcome in countries with more liberal policies1 but it is still illegal to import marijuana and its derivatives into the US.
Given that synthetic and botanical extract products can be approved via the 505(b)(2) pathway, one of the most important considerations is how to leverage existing information in the literature to reduce the size and scope of the development program.
Cannabis-extract Products
The FDA has never approved botanical cannabis or any product derived from it. This is because no-one has ever asked the FDA to approve a cannabis-derived product via the correct regulatory procedures. Readers of this blog are sophisticated enough to know that as per statute, the FDA can only approve a product after reviewing an application that provides sufficient evidence of efficacy and safety. So despite all the attention from media and patient advocacy groups, cannabinoid-based products must follow the same rules as any other drug product.
Sativex® (nabiximols oral mucosal spray; GW Pharmaceuticals plc) was approved in the UK for moderate to severe spasticity due to multiple sclerosis in 2010 making it the first approved cannabis-based prescription medicine in the world. Sativex contains tetrahydrocannabinol (THC), cannabadiol (CBD), other cannabinoids and non-cannabinoids. It is now also approved in other countries but not the US. GW Pharmaceuticals, a British company, reported in 2014 that it has been granted fast track designation for Sativex by the FDA for the treatment of pain in patients with advanced cancer who experience inadequate analgesia during optimized chronic opioid therapy. Mixed trial results in this population have apparently slowed progress.
GW pharmaceuticals is also conducting multiple Phase 3 clinical trials for a CBD-based product (Epidiolex®) for several treatment-resistant childhood epilepsy syndromes. The company reported that it expects to submit an NDA for Epidiolex by the end of June 2017.
Synthetic Cannabinoids
Several approved products are synthetic derivatives of THC, the primary psychoactive substance in marijuana, as shown in the table below.
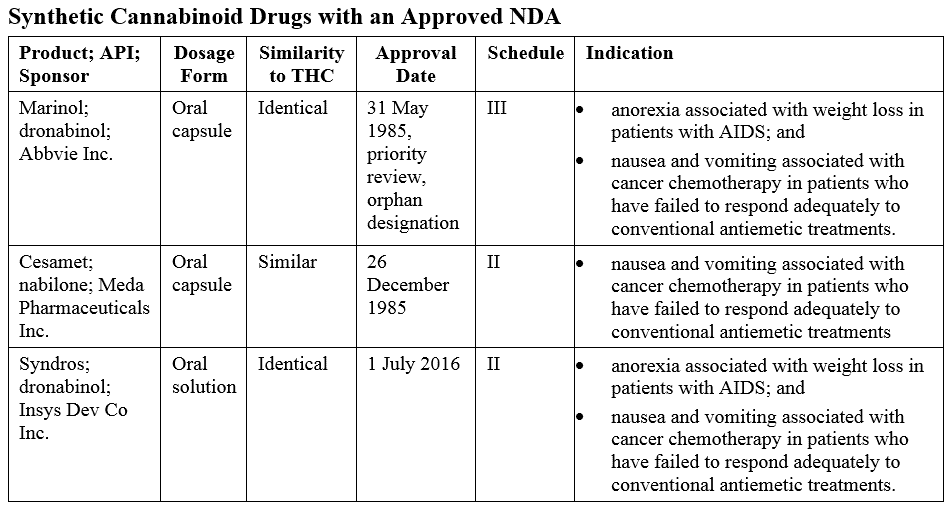
Syndros was approved via the 505(b)(2) pathway as it is a new dosage form of dronabinol.
Regulatory Background
As controlled substances, cannabinoids are subject to regulation by the FDA and the Drug Enforcement Agency (DEA). After a product has been approved, the FDA notifies the DEA. The Department of Health and Human Services (HHS), of which the FDA is an agency of, then provides the DEA with a scientific and medical evaluation and scheduling recommendation. The DEA then determines the schedule for the product. The schedule affects the controls necessary for prescribing, supplying, or storing the product, with a lower number being the most restrictive.
Marijuana is currently classified as a schedule I substance of the Controlled Substances Act which means it lacks an established medical use (from a regulatory perspective) and has a high potential for abuse. If a medical use can be adequately demonstrated, the Sponsor of a potential cannabis-derived drug would petition to have it moved to a less restrictive schedule depending on the DEA’s assessment of its potential for psychological or physiological dependence.
Unimed Pharmaceuticals Inc., the original Sponsor of Marinol, petitioned the Agency to reschedule from schedule II to III. In 1999, this was granted because of “the findings of the DEA that the difficulty of separating dronabinol from the sesame oil formulation and the delayed onset of behavioral effects due to oral route administration supported a lower abuse potential of Marinol as compared to substances in schedule II,” 64FR 35928. Unimed then submitted a supplemental NDA as Changes Being Effected and the Marinol labeling was updated to reflect the new schedule.
Scheduling for Syndros
Syndros was approved on 1 July 2016. The Sponsor has been unable to market the product until a schedule was determined by the DEA and the labeling updated to reflect this schedule. Last week, the DEA issued an interim final rule placing FDA-approved oral solution products containing dronabinol in schedule II.
The difference between the scheduling of Marinol and Syndros came down to the dosage form. Both products share the same indications, pharmacology, and potential for abuse in human ‘drug liking’ studies. But Syndros was determined to be more amenable to extraction and subsequent abuse. The interim final rule stated that:
“HHS indicated that the formulation of Syndros (oral solution) is easier to abuse than Marinol because this liquid formulation can be manipulated to produce concentrated extracts of dronabinol for abuse by inhalation (smoking or vaping) or through other routes of administration. Because of the large amount of dronabinol in Syndros oral solution it has a greater potential for extraction than Marinol and thus has a greater abuse potential. Based on the data from in vitro studies conducted by the Sponsor, the large amount of dronabinol in the Syndros formulation, its pharmacokinetics upon oral administration, and its contribution to marijuana psychoactivity, HHS stated that the abuse potential of the dronabinol oral solution is similar to that of other THC containing products such as concentrates, infused edibles and drinks. Similar to these THC containing products, Syndros oral solution can be easily manipulated to other forms that can be easily abused through inhalation and oral routes of administration.” 82 FR 14815
The DEA’s interim final rule became effective on 23 March 2017 and is now in a 30-day comment period. Of note, although HHS requested DEA to place all dronabinol dosage forms in schedule II, the interim final rule does not include a rescheduling of Marinol and will not apply to other dosage forms being developed such as Axim Biotechnologies Inc.’s proposed dronabinol controlled-release chewing gum.
Getting the FDAs Stamp of Approval
A Sponsor of a proposed cannabis-extract product will need to meet the quality requirements for a botanical drug, as described in the December 2016 revised FDA Guidance for Industry: Botanical Drug Development, in addition to the usual safety and efficacy requirements. Ensuring that the therapeutic effect between batches is consistent can be more challenging for a botanical product, especially when one considers that there are hundreds of compounds in cannabis, some with modulatory effects on others. The purity and potency of the proposed product must be demonstrated as part of the approval process. There has only ever been one botanical drug product, according to the definition in the Botanical Guidance, that has been approved for marketing as a prescription drug (sinecatechins, Veregen®)2.
For a synthetic cannabinoid that relies on the FDAs findings of safety and efficacy, it is worth keeping in mind that the FDA’s standards have changed since the approval of Marinol and Cesamet®. Additional studies may be required even if the proposed product only differs from an approved product in the dosage form but has the same active pharmaceutical ingredient and the same indication. This was the case in the 2016 approval of Syndros. Although the full review details have not yet been released, additional toxicity studies and pediatric studies are required as part of a post-marketing commitment. These studies include a 3-month repeat-dose toxicity study, a pre/postnatal developmental toxicology study, and pharmacokinetic and a tolerability/efficacy studies in pediatric cancer patients.
Of course, an abuse liability assessment will be required. This will be based on drug liking studies. These studies are designed to assess the abuse potential for a product, often compared to an existing product, in recreational drug users.
Premier Consulting has worked with several Sponsors wishing to develop cannabinoid products and has significant experience with ‘drug liking’ studies required for approval. To learn more about the development program required for approval of your product, contact us.
Author:
Angela Drew, PhD
Product Ideation Consultant